Mauve[1][2][3] 可用于构建多个小型基因组的比对,可以解释进化关系,也能够快速有效分析基因组之间有无大片段序列重排现象,及基因组间共线性是否良好,以及是否存在局部共线区( LCBs ),这些分析都为比较基因组学的研究和全基因组进化动力学的研究提供了基础。
# 安装
java -version
openjdk version "1.8.0-292"OpenJDK Runtime Environment (build 1.8.0-292-b10)
OpenJDK 64-Bit Server VM (build 25.71-b10, mixed mode)
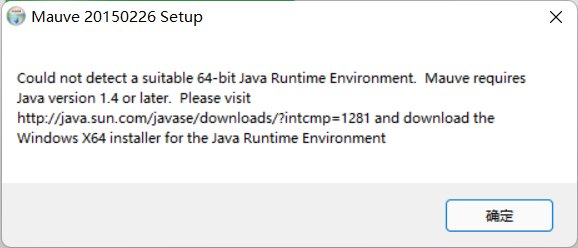
若没有安装或未正确配置环境变量会有警告,然后自动停止安装。
![fig:01]()
如果系统包含多个版本的 Java 请暂且将 Java8 的环境变量优先级调高。
![fig:02]()
选择适合自己平台的包。
# 运行
Mauve 的 GUI 由 Mauve.jar 包提供,算法核心是 mauveAligner 和 progressiveMauve 。因此我们除了双击桌面快捷方式或者 jar 包的方式打开,还可以直接用命令打开[4](请先将 Mauve 安装路径加入到环境变量):
javaw -jar -Xmx1000m Mauve.jar |
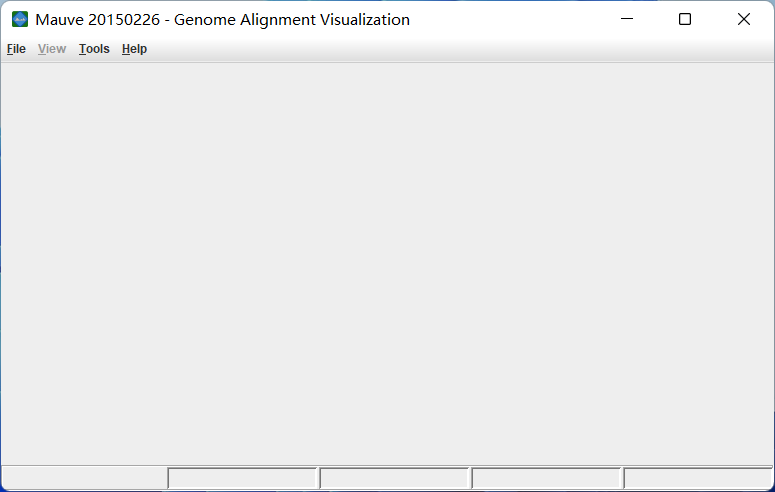
若能打开如下界面则安装成功:
# 使用
# 序列比对
# 图形界面
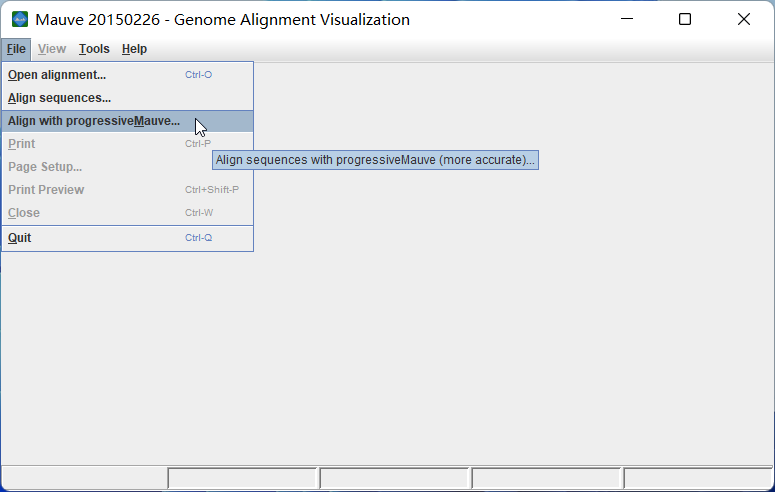
点击 File -> Align with progressiveMauve :
点击 Add Sequence... :
选择想要比对的序列文件(可以全选),这里用到的是 lactobacillus gasseri 全基因组的前三株[5]:
点击 Output 右边的 ... ,输入保存文件名(可以是任意名称,且不需要指定拓展名):
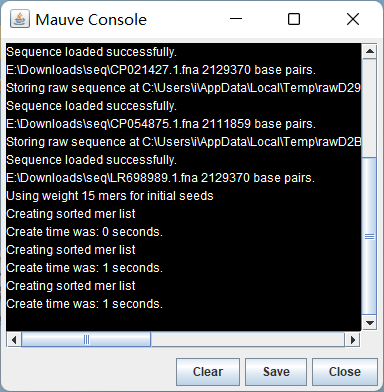
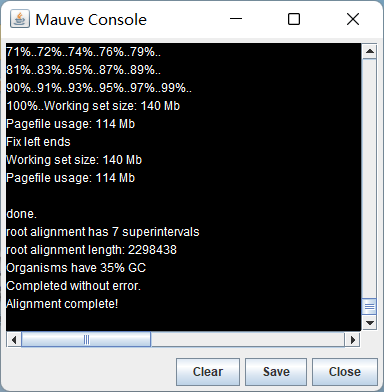
Mauve Console 会有运行日志,此时不要关闭任何窗口:
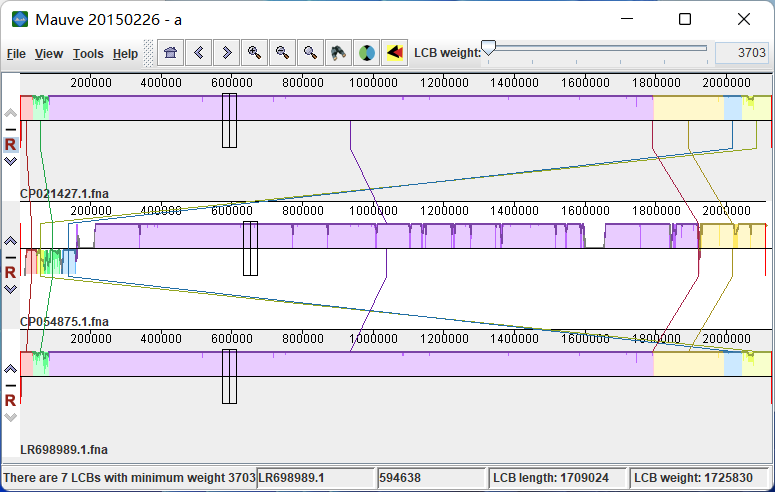
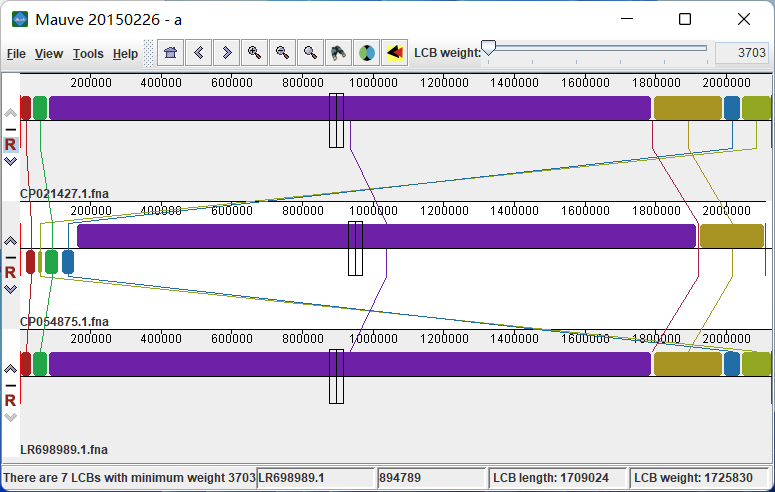
当看到 Mauve Console 最后一行是 Alignment Complete! 且弹出图像[6],即运行结束:
生成的无拓展名的 a 包含着图像的信息:
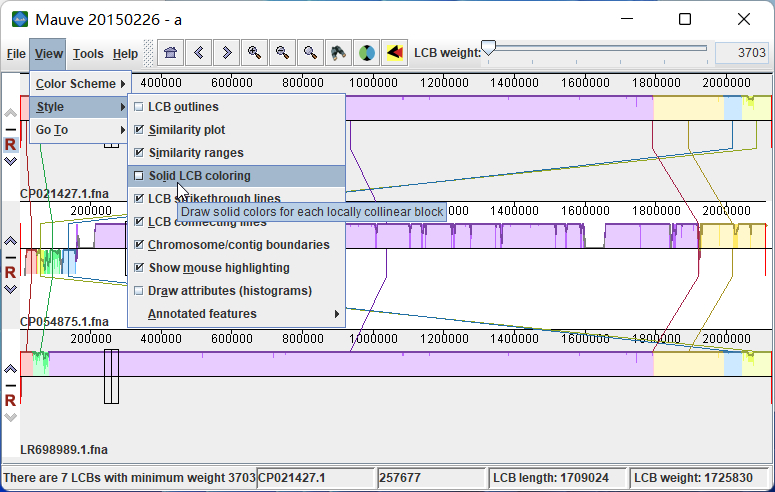
可以设置图像的风格,比如色块颜色加深:
图像可以导出,代替截图:
还可以导出 PDF,配合 pdf2svg 和 Visio 制作矢量图。
# 命令行
progressiveMauve 比 mauveAligner 稍微精确些,这里只用 progressiveMauve :
progressiveMauve --output=./a.xmfa \ | ||
--output-guide-tree=./a.tree \ | ||
--backbone-output=./a.backbone \ | ||
./CP021427.1.fna \ | ||
./CP054875.1.fna \ | ||
./LR698989.1.fna |
# 文件格式
详细的文件格式
.alignment格式:以 eXtended Multi-FastA 格式存储 Mauve 产生的比对数据。.xmfa格式:存储了多个共线性块的比对情况,不同共线性块以 = 分割。每个共线性块中,一个基因组有一条对应的 fasta 格式的序列。其中,定义行给出这条序列位于基因组的位置和方向(正负链)。这些共线性块共同组成了基因组比对结果。.bbcols格式:存储了比对中发现的 genomic islands,以 tab 键分割。Island 指的是比对中一部分基因组有,而另一部分基因组没有的区域。一个 island 由一个序列的基因组坐标定义,其中另一个基因组在比对的那部分中包含长度为 n 或更长的缺口。缺口的长度 n 可以人为定义。.backbone格式:存储了在所有基因组中都是保守的比对区域(片段的起始位点)。SNP文件:记录了 SNP 模式(按照输入顺序排序)以及 SNP 在每个基因组中的位置。orthologs文件:
# 原始 Mauve 算法和 progressive Mauve 算法[7]
原始 Mauve 算法的优势:比较适合 亲缘相近 物种
原始 Mauve 算法的劣势:
- 不比对部分基因组共享的大区域
- 识别不到部分基因组共享的重排区域
- 为了准确估计基因组重排,必须人工调整最小 LCB 权重
- 很难比对经常出现片段重复的基因组
progressive Mauve 算法的优势:
- 能够比对更多的基因组
- 能够比对分歧更大的基因组,核苷酸相似性可以低至 50%
- 不需要人工调整比对打分参数
- 可以比对
泛基因组 - 准确度更高
progressive Mauve 算法的劣势:
- 很慢
- 很耗内存